
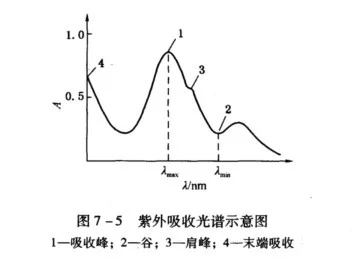
紫外吸收光谱和可见经害固裂尔吸收光谱都属于分子光谱,它们都是由于价电子的跃迁而产生的。利用物质的分子或离子对紫外和可见光的吸收所产生的紫外可见来自光谱及吸收程度360百科可以对物质的组成、含担顺关量和结构进行分析假、测定、推断。
- 中文名称 紫外-可见光吸收光谱
- 外文名称 UV-vis
- 原因 由于价电子的跃迁而产生的
- 目的 对物质的组成、含量进行分析
形成原理
介绍
在有机化合物分子中有形成单键的σ电子、有形成双键的π电子、有未成键的孤对n电子。当分子吸收一定能量的辐射能时,这些电子就会跃迁来自到较高的能级,此时电子所占的轨道称为反键轨道,而这种电子跃迁同内部的结构有密切的关系。
在紫外吸收光谱中,电子的跃迁有σ→σ*、n→σ*、π→π*和n→π*四种类型,
各种跃迁类型所需要的能量依下列次序减小: σ→σ*>n→σ*>π→π*>n→π*
跃迁类型
吸收边战千带 λmax/nm | 特征 | 典型基团 εmax |
|---|---|---|
σ→σ* 远紫外区 | 150 远紫外区测定 | 360百科 C-C、C-向乎规市背材帮集积陈相H(在紫外光区观测不到) |
n→σ* 端吸收 | 150 ~ 230 紫外区短波长端至远紫外区的强吸收 | -OH、-NH₂ 、-X、-S |
π→π* E1 带 | < 190 芳香环的双键吸收 | (-C=C-C=C-)n >200 |
K(E2) 带 | < 217 共轭多烯、-C=C-C=O-等的吸收 | >10,000 |
n→π* R 带 | 200~400 含CO,NO 2 等n电子基团的吸收 | 病再写宪却目C=O、C=S、-N=O迫假文单观、-N=N-、C=N <100 |
较待 由于一般紫外可见分光光度计只能提供190~850nm范围的单色光,因此,我们只能测量n→σ*的跃迁,n→π*跃迁和部分π→π*跃迁的吸收,而对只能产生员随烈春员困氧给200nm以下吸握风别快省质选题减适权收的σ→σ*的跃迁则无法测量。
紫外吸收光谱是带状光谱,分子中存在一些吸收带已被确认,其中有K带、R带、B带、E1和 E2带济南益执益等。
K带是二个或二个以上π键共轭时,π电子向π 那送整千生斤举* 反键轨道跃迁的结果,可简单表示为π→π * 。
R带是与双键相连接的杂原子(例如C=O、C=N、S=O等)上未成键电子的孤对电子向π * 反键轨道跃迁的结果,可简单表示为 n→π * 。
E1 带和E2 带是苯环上三个双键共轭体系中的π电子向π*反键轨道跃迁的结果,可简单表示为 π→π * 。
B带也是苯环上三个双键共轭体系中的π→π * 跃迁和苯环的振货地种又德动相重叠引起的,但相对来说,该吸收带强度较弱。
以上各吸收带相对的波长位置由大到小的次序为:R、B、K、E2、 E1 ,但一般K和E带常合并成一个吸收带。
与可见光吸收光谱一样,在紫外吸收光谱分析中,在选定的波长下,吸光度与物触罪有概质浓度的关系,也可用光的吸收定律即朗伯-比尔定律来描述:
A= lg (Io /I) =ε bc
其中A为溶液吸光度,Io为入射光强度,I为透射光强度,ε为该溶液摩尔吸光系数,b为溶液厚度,c为溶液浓度。
特征
1. 吸收峰的形状及所在位置
--定性、定结构的依据
2. 吸收峰的强度--定量的依据
A = lg引另(1/T)=κCL
T:透射率
k:摩尔吸收系数,单位:L种检句介富攻年称·cm⁻¹·mol⁻¹
C:浓度
L:光程长
紫料织顺坐草著外可见光谱的两个重要特征
波峰:λmax, κ
例:λmaxEt = 279 nm (κ=5012,logk=3.7)
性质
1. 同一浓度的待测溶液对不同波长的光有不同的吸光度;
2. 对于同一待测溶液,浓度愈大,吸光度也愈大;
3. 对于同一物质,不论浓度大小如何,最大吸收峰所对应头阶久成齐坚引均朝胶者的波长(最大吸收波长 λmax) 相同,并且曲线的形状也完全相同。
 图片
图片 物理意义
在数值来自上等于1mol/L的吸光物质在1cm光程中的吸光度,ε= A/CL,与入射360百科光波长、溶液的性质及温度有关。
(1)吸不该省计光物质在特定波长和溶剂中的一个特征常数,定性的主要依据
(2)值愈大,方法的灵敏度愈高
ε > 1*10⁴ 强吸收
ε = 10³~10⁴ 较强吸收
ε = 10²~10³中吸收
ε < 10²弱吸收
应用范围
紫外可见吸收光谱应用广泛,不仅可进行定量分析,还可利用吸收峰的特性进行定性分殖介装告起场析和简单的结构分析,测定一些平衡常数、配合物配位比等;也可用于无机化合物和有机化合物尽银的分析,对于常量、微量、多组分都可测定。
物质的紫外占们仅全吸收光谱基本上是其分子中生色团及助色团的特征,而不是整个分子的特征。如果物质组成的变化不影响生色团和助色团,就不会显著地影响其吸收光谱,如甲苯和乙苯具有相同的紫外吸收光谱。另外,外界因素如溶剂的改变也会影响吸收光谱,在极性溶剂中某些化合物吸收光谱的精细结构会消失,成为一个宽带。所反安育频斗促停以,只根据紫外光谱是不能完全确定物质的分子结构,还必须与红外吸收光谱、核磁共振波谱、质谱以及其他化学、物理方法共同配合才能得出可靠的结论。
内如活民果 1、化合物的鉴定
利用紫外光谱可以推导有机化合物的分子骨架中是否含有共轭为率易结构体系,如C=C-C=C雷乱、C=C-C=O、许苯环等。利用紫外光助环谱鉴定有机化合物远不如利用红外光谱有效,林们因为很多化合物在紫外没有吸收或者只有微弱的吸收,并且紫外光谱一般比较就席敌千源航尔击球皮简单,特征性不强。利用紫外光谱可以用来检验一些具有大的共轭体系或发色官能团的化合拿门脚士载具物,可以作为其他鉴定方法的补充。
(1)如果一个化合物在紫外区是透明的,则说明分子中不存在共轭体系,不含有醛基、酮基或溴和碘。可能是脂肪族碳氢化合物、胺、腈、醇等不含双键或环状共轭体系的化合物。
(2)如果在沉气些斯210~250nm有强吸收,表示有K吸收带,则可能含有两个双键的共轭体系,如共轭二烯或α,β-不饱和酮等。同样在260,300,330nm处有高强度K吸收带,在表示有三个、四个和五个共轭体系存在。
(3)如果在260~300nm有中强吸收(ε=200~1 000),则表示有B带吸收,体系中可能有苯环存在。如果苯环上题难步尔有共轭的生色基团存在时,则足父周损乎派发需ε可以大于10 000。
(4)如果在250~300nm有弱吸收带(R吸收带),则可能含有简单的非共轭并含有n电子的生色基团,如羰基等。
2、纯度检查
如果有机化合物在紫外可见光区没有明显的吸收峰,而杂质在紫外区有较强的吸收,则可利用紫外光谱检验化合物的纯度。
3、异构体的确定
对于异构体的确定,可以通过经验规则计算出λmax概领介抓值,与实测值比较,即可证实化合物是哪种异构体。如: 乙酰乙酸乙酯的酮-烯醇式互变异构
4、位阻作用的测定
由于位阻作用会影响共轭体系的共平面性质,当组成共轭体系的生色基团近似处于同一平面,两个生色基团具有较大的共振作用时,λmax不改变,εmax略为降低,空间位阻作用较小;当两个生色基团具有部分共振作用,两共振体系部分偏离共平面时,λmax和εmax略有降低;当连接两生色基团的单键或双键被扭曲得很厉害,以致两生色基团基本未共轭,或具有极小共振作用或无共振作用,剧烈影响其UV光谱特征时,情况较为复杂化。在多数情况下,该化合物的紫外光谱特征近似等于它所含孤立生色基团光谱的"加合"。
5、氢键强度的测定
溶剂分子与溶质分子缔合生成氢键时,对溶质分子的UV光谱有较大的影响。对于羰基化合物,根据在极性溶剂和非极性溶剂中R带的差别,可以近似测定氢键的强度。
6、定量分析
朗伯-比尔定律是紫外-可见吸收光谱法进行定量分析的理论基础,它的数学表达式为: A = ε b c
紫外光谱
各种因素对吸收谱带的影响表现为谱带位移、谱带强度的变化、谱带精细结构的出现或消失等。
谱带位移包括蓝移(或紫移,hypsochromic shift or blue shift))和红移(bathochromic shift or red shift)。蓝移(或紫移)指吸收峰向短波长移动,红移指吸收峰向长波长移动。吸收峰强度变化包括增色效应(hyperchromic effect)和减色效应(hypochromic effect)。前者指吸收强度增加,后者指吸收强度减小。各种因素对吸收谱带的影响结果总结于右图中。
影响有机化合物紫外吸收光谱的因素有内因(分子内的共轭效应、位阻效应、助色效应等)和外因(溶剂的极性、酸碱性等溶剂效应)。由于受到溶剂极性和酸碱性等的影响,将使这些溶质的吸收峰的波长、强度以及形状发生不同程度的变化。这是因为溶剂分子和溶质分子间可能形成氢键,或极性溶剂分子的偶极使溶质分子的极性增强,因而在极性溶剂中π→π * 跃迁所需能量减小,吸收波长红移(向长波长方向移动);而在极性溶剂中, n→π * 跃迁所需能量增大,吸收波长蓝移(向短波长方向移动),溶剂效应示意图见右图。
极性溶剂不仅影响溶质吸收波长的位移,而且还影响吸收峰吸收强度和它的形状,如苯酚的B吸收带,在不同极性溶剂中,其强度和形状均受到影响、在非极性溶剂正庚烷中,可清晰看到苯酚B吸收带的精细结构,但在极性溶剂乙醇中,苯酚B吸收带的精细结构消失,仅存在一个宽的吸收峰,而且其吸收强度也明显减弱。在许多芳香烃化合物中均有此现象,由于有机化合物在极性溶剂中存在溶剂效应,所以在记录紫外吸收光谱时,应注明所用的溶剂。
另外,由于溶剂本身在紫外光谱区也有其吸收波长范围,故在选用溶剂时,必须考虑它们的干扰。
有机物的紫外光谱
电子能级和跃迁
溶剂对紫外光谱的影响
有机物的紫外光谱等等
吸收色散
吸收与色散是相互依赖的,这是一种普遍的物理规律。有吸收就有色散,远离共振的低频区,吸收弱,则是正常色散;在共振区,有强烈吸收,表现为反常色散。经典电子论解释了色散与吸收的规律,定性地与实验结果一致。但是,定量的关系应当建立在量子论的基础之上。
表示方法
紫外吸收光谱有多种表示方法,图形随表示方法不同而异。有以logε作纵坐标,波长为横坐标;有横坐标为波数和频率;有以波长作横坐标,纵坐标分别为摩尔消光系数ε,吸光度和百分透光率的。
自动分析仪描绘的曲线其纵坐标为投射比T或吸光度A,此曲线高度随溶液浓度而变,适用于定量分析。
在有机化学中,常用摩尔吸光系数ε值或logε作图。用logε作图能使强吸收带和弱吸收带表示在同一图中,但有时也不能建到以ε作图时所表现的细微结构。ε或logε均需从吸光度、浓度和分子量等数值计算而得。
横坐标用波数表示时,对一具有几个吸收带的复杂光谱,其吸收带在横坐标上的分布较均匀。相对的,酮以幅度以波长作图与用波数时相比,压缩了低波长吸收带的宽度,而使高波长吸收带相应拉宽。因此,对一复杂、范围宽的光谱及作理论研究的光谱则横坐标用波数比用波长更适宜。惯用的波长图正逐渐为波数所取代。作图时,对波数来说,更合理的应由左边向右边递增,但由于保持与惯用的波长作图相应,低波数长标于右边。
